
When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. You’re not just saving money-you’re trusting that the FDA has made sure it’s just as safe and effective. But how does the FDA actually make sure a generic drug made in India, China, or Ohio meets the same standards as the original? It’s not luck. It’s a system built on science, inspections, and constant monitoring.
The Legal Foundation: Hatch-Waxman and the ANDA Pathway
The modern system for approving generic drugs started in 1984 with the Hatch-Waxman Act. Before this, companies had to run full clinical trials to prove a generic drug was safe and effective-just like the brand-name version. That made generics too expensive and slow to bring to market. Hatch-Waxman changed that. It created the Abbreviated New Drug Application (ANDA), which lets manufacturers skip costly clinical trials if they can prove their drug is bioequivalent to the brand-name drug.Bioequivalence means the generic delivers the same amount of active ingredient into the bloodstream at the same rate as the original. The FDA requires this range to be 90-110%. That’s not a guess. It’s based on decades of pharmacokinetic studies. If a generic meets this, it’s considered therapeutically equivalent. No extra trials needed. But here’s the catch: skipping clinical trials doesn’t mean skipping scrutiny. The FDA still digs deep into every part of the manufacturing process.
How the FDA Reviews Generic Drug Applications
Every ANDA goes through a multi-layered review by the Center for Drug Evaluation and Research (CDER), specifically its Office of Generic Drugs (OGD). The review isn’t just about the numbers-it’s about the entire story of how the drug is made.First, the manufacturer must prove pharmaceutical equivalence. That means the generic has the same active ingredient, strength, dosage form, and route of administration as the brand. Then comes bioequivalence data-usually from studies with healthy volunteers. But that’s only half the story. The FDA also checks the labeling to make sure it matches the brand’s exactly. No hidden warnings. No misleading directions.
Then comes the inspection. Before approval, the FDA inspects every facility involved: where the active ingredient is made, where it’s mixed, where it’s packaged. These aren’t routine visits. They’re full audits. Inspectors look for compliance with Current Good Manufacturing Practices (cGMP). If there’s even one major violation, the application gets a Complete Response Letter-essentially a rejection-and the company has to fix it before resubmitting. Many applications go through multiple cycles, each taking months. The average review time used to be over two years. Today, thanks to user fees from GDUFA, 95% of standard ANDAs are reviewed within 10 months.
Manufacturing Rules: cGMP and Quality Control
The FDA doesn’t just check paperwork. They make sure the factory operates like a precision machine. Every step must be documented, controlled, and repeatable. There are three non-negotiable pillars of quality control:- Raw material control: Every batch of active pharmaceutical ingredient (API) must be tested for purity, potency, and contamination. Suppliers are tracked back to their source. If a batch fails, the entire production run is rejected.
- Process controls: Every machine setting, temperature, mixing time, and pressure is recorded. If a parameter drifts outside the approved range, the batch is quarantined. There’s no room for improvisation.
- Final product testing: Finished pills are tested for dissolution, uniformity, and stability. They’re stored under different conditions to simulate real-world shelf life. If the drug breaks down too fast, it’s pulled.
These aren’t suggestions. They’re legal requirements. A single violation can trigger a warning letter, import alert, or even a shutdown. In 2019, the FDA found quality issues in 15% of foreign manufacturing facilities-nearly double the rate of domestic ones. That’s why inspections have shifted focus.
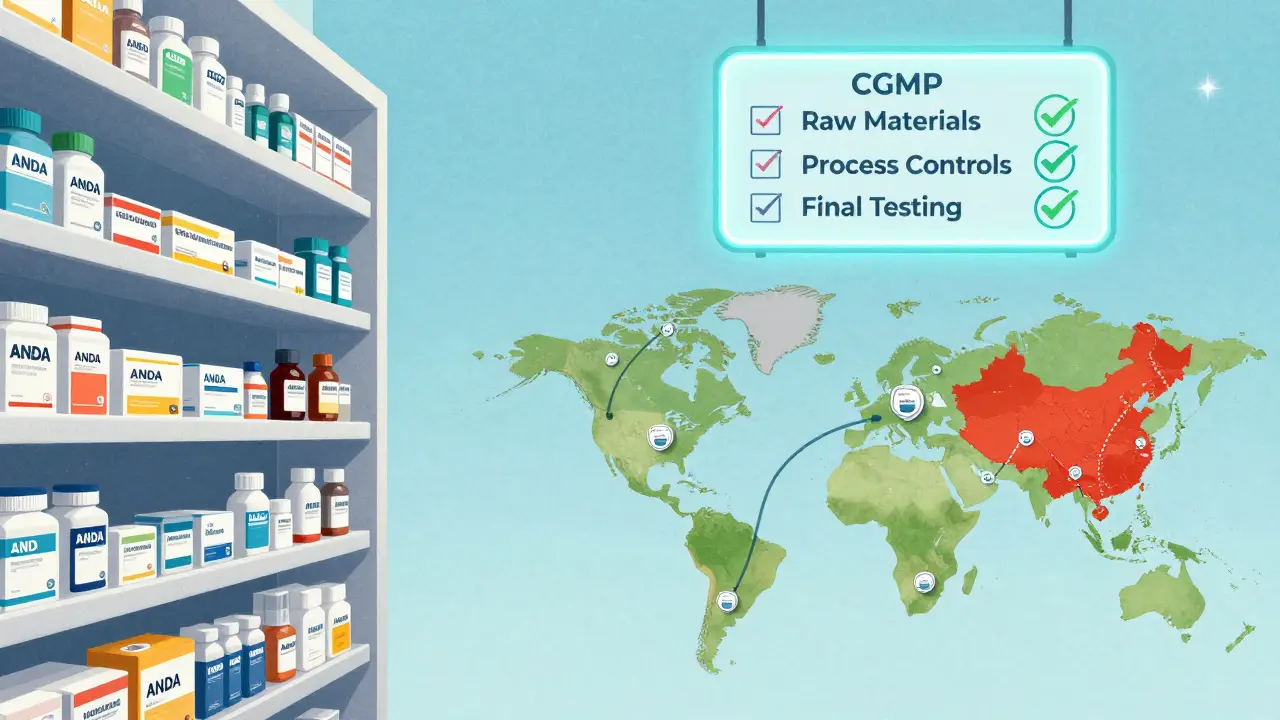
Global Inspections: The Foreign Factory Challenge
More than 80% of the active ingredients in U.S. generic drugs come from overseas. India and China are the biggest suppliers. That means the FDA can’t just inspect U.S. plants. They have to fly halfway around the world.In 2021, the FDA conducted 1,082 inspections globally. By 2025, they’re targeting 1,500-up nearly 40% in just four years. This push is funded by GDUFA III, which allocated $1.1 billion through 2027 specifically to strengthen foreign oversight. The agency now uses a risk-based system. Factories with past violations, poor inspection history, or complex products get priority. Some get inspected every two years. Others, if they’ve been flawless for a decade, might wait longer.
But it’s not just about frequency. The FDA now uses real-time data to spot red flags. If a company’s export records show sudden spikes in shipments to the U.S. after a quality issue, inspectors can jump on it. They also collaborate with regulators in India, China, and the EU to share inspection reports and avoid duplication.
Post-Market Surveillance: Watching After Approval
Approval isn’t the finish line. It’s the starting line. The FDA keeps watching-even after the drug hits shelves.Doctors, pharmacists, and patients report side effects through MedWatch. In 2023, the system processed over 1.3 million reports. The Division of Clinical Safety and Surveillance digs into these, looking for patterns. If a generic drug suddenly shows more reports of dizziness or liver problems than the brand-name version, the FDA investigates. Sometimes it’s a manufacturing change. Sometimes it’s a batch issue. Sometimes it’s just coincidence.
If a problem is confirmed, the FDA can take action. That might mean a voluntary recall, a label update, or a “Dear Healthcare Provider” letter warning doctors to monitor patients more closely. In 2022, the FDA issued 17 safety alerts for generic drugs-most tied to impurities or packaging defects.
The agency also monitors drug shortages. If a single factory producing a critical generic (like insulin or blood pressure meds) shuts down, the FDA can fast-track approval of another manufacturer to fill the gap. That’s happened dozens of times in the last five years.

Why This System Works-And Where It Still Struggles
The results speak for themselves. Generic drugs make up 90% of prescriptions in the U.S. but cost only 23% of total drug spending. They saved the healthcare system $313 billion in 2022 alone.But the system isn’t perfect. Some manufacturers still cut corners. In 2020, a foreign plant was found to be falsifying test data on a blood thinner. The FDA pulled the drug from the market, but not before thousands of patients received potentially unsafe doses.
Another challenge: complex generics. Inhalers, injectables, and topical creams are harder to copy than a simple tablet. The FDA has launched the Complex Generic Drug Products Initiative to create specific guidance for these. But progress is slow. Some drugs still take years to get approved because the science is still catching up.
Still, the FDA’s approach is the most comprehensive in the world. No other country combines pre-approval reviews, mandatory inspections, real-time surveillance, and global coordination the way the U.S. does.
What You Can Do as a Patient
You don’t need to be a scientist to protect yourself. Here’s what you can do:- Check if your generic is FDA-approved. Use the Drugs@FDA database. Look for the “ANDA” number on the label.
- Report side effects. If you notice something new-rash, dizziness, nausea-tell your doctor and file a report with MedWatch.
- Don’t assume all generics are identical. If you switch brands and feel different, talk to your pharmacist. Sometimes small differences in fillers or coatings affect absorption.
The bottom line: generic drugs are safe because the FDA treats them like they’re brand-name drugs. They don’t get a pass. They get a second look-every time.
Are generic drugs really as safe as brand-name drugs?
Yes. The FDA requires generic drugs to have the same active ingredient, strength, dosage form, and bioequivalence as the brand-name version. They must meet the same strict manufacturing standards (cGMP) and undergo the same quality testing. Studies show they work the same way in the body and have identical safety profiles.
How does the FDA know if a generic drug is made safely overseas?
The FDA inspects foreign manufacturing facilities just like U.S. ones. Since 2012, the agency has increased foreign inspections by over 150%. Under GDUFA III, the goal is to inspect all high-risk foreign sites every two years. The FDA also shares inspection data with international regulators and uses real-time supply chain data to flag suspicious activity.
What happens if a generic drug causes side effects after approval?
The FDA monitors all approved drugs through MedWatch, receiving over a million reports annually. If a pattern emerges-like more reports of liver damage or allergic reactions-the agency investigates. If a safety issue is confirmed, they can issue a recall, update the drug label, or send a warning letter to doctors. This system has caught issues with impurities, packaging errors, and inconsistent dosing.
Why do some people feel different on a generic version?
While the active ingredient is identical, generics can use different inactive ingredients-like fillers, dyes, or coatings. For most people, this makes no difference. But for those with allergies or very sensitive conditions (like epilepsy or thyroid disorders), even small changes can affect how the drug is absorbed. If you notice a change after switching, talk to your doctor. You may need to stick with one brand.
How long does it take for the FDA to approve a generic drug?
The legal deadline is 180 days, but the average review time is now about 10 months. Before 2012, it took nearly three years. Thanks to the Generic Drug User Fee Amendments (GDUFA), the FDA hired more reviewers and streamlined the process. Complex drugs or those with manufacturing issues still take longer, sometimes over a year.
Can the FDA shut down a generic drug factory?
Yes. If a facility repeatedly violates cGMP standards, the FDA can issue a warning letter, block imports, or even pursue criminal charges. In 2017, a major Indian manufacturer was banned from exporting to the U.S. after multiple inspections found falsified data. The FDA has the authority to remove any drug from the market if it’s deemed unsafe or improperly made.
Matt Davies
This is the kind of system that makes me proud to live in a country that still takes public health seriously. 🌟 The FDA doesn’t just rubber-stamp pills-they chase down every impurity, every dodgy factory, every falsified report like it’s their personal mission. And yeah, it’s expensive, it’s tedious, and yes, some foreign plants still slip through-but the fact that 90% of our meds are safe and cheap? That’s not magic. That’s grit.
Jedidiah Massey
It’s fascinating how the ANDA pathway represents a triumph of regulatory economics-bioequivalence thresholds (90–110%) are grounded in pharmacokinetic variance models derived from log-normal distributions of Cmax and AUC, not arbitrary benchmarks. The GDUFA fee structure has been instrumental in reducing review latency, though the marginal cost of inspecting high-risk foreign facilities remains undercapitalized relative to the scale of global API sourcing. Still, the data integrity protocols are among the most rigorous in the world.
Dominic Suyo
Let’s be real-the FDA is a joke. They inspect one factory in India, get a fancy PowerPoint, and then act like everything’s perfect. Meanwhile, 15% of foreign sites have major violations? That’s not oversight, that’s negligence dressed up in lab coats. And don’t even get me started on how they ‘monitor’ post-market side effects. You think some guy in Ohio reporting dizziness after switching generics actually gets looked at? Nah. It’s all PR.
Vicki Belcher
Y’all need to stop panicking about generics 😊 I switched from brand-name blood pressure med to generic last year and haven’t had a single issue. The FDA’s system is actually insane in the best way-like, they test the pills under heat, humidity, light, you name it. And if something’s off? They pull it. No drama. Just science. 🤓💊
Mark Able
Wait, so if I’m taking a generic version of my antidepressant and I suddenly feel like a zombie, it’s NOT the drug-it’s the filler? That’s wild. Can someone tell me how to find out what’s in my pills? Like, the exact inactive ingredients? I’m not asking for a PhD thesis-I just want to know if it’s got gluten or dye that’s making me feel like I’m drowning in slow motion.
Dorine Anthony
Just read this whole thing. Honestly? I didn’t know half of this. I always assumed generics were just cheap knockoffs. Turns out they’re like the unsung heroes of healthcare. Respect to the FDA inspectors flying halfway across the world to check factories. I’m gonna start checking my pill bottles for the ANDA number now.
William Storrs
Look, I know it sounds boring-but this is the stuff that keeps you alive. The FDA doesn’t get enough credit. They’re the quiet ones in the back, making sure your insulin doesn’t turn to sludge in your fridge. You don’t need to thank them. Just keep taking your meds. And if you’re feeling weird after a switch? Talk to your pharmacist. That’s your power move.
James Stearns
While the regulatory framework presented herein is ostensibly robust, it remains fundamentally compromised by structural underfunding and geopolitical dependency. The reliance upon foreign manufacturing entities-particularly those in jurisdictions with opaque governance structures-constitutes a critical vulnerability. The assertion that 90-110% bioequivalence ensures therapeutic equivalence is a statistical fallacy when applied to narrow-therapeutic-index agents. One must question the ethical integrity of a system that permits such risk.
Nina Stacey
My grandma takes five different generics and swears she feels different every time she switches brands. She says one makes her dizzy, another makes her stomach hurt, and the third one? She says it tastes weird. I told her it’s all the same chemical, but she’s not buying it. Maybe she’s right. Maybe the little things matter more than we think. I’m gonna start keeping a journal. Like, pill name, date, how I felt. Maybe that’s the real oversight we’re missing.
Carolyn Benson
Isn’t it ironic? We outsource the production of our most vital medicines to nations with questionable labor and environmental standards, then celebrate the FDA for ‘inspecting’ them like they’re visiting a school science fair. We demand safety, yet we refuse to pay for it. We want cheap pills but act shocked when someone cuts corners. The system isn’t broken-it’s designed this way. Capitalism doesn’t care if you live or die. It just cares if the paperwork is signed.
mark shortus
THEY FOUND FALSIFIED DATA ON A BLOOD THINNER AND DIDN’T RECALL IT FAST ENOUGH. PEOPLE DIED. AND NOW WE’RE CELEBRATING THE FDA LIKE THEY’RE SUPERHEROES?? 😭 THIS SYSTEM IS A HOUSE OF CARDS. ONE BAD BATCH. ONE CORRUPT LAB. ONE OVERWORKED INSPECTOR. AND WHAM. YOU’RE DEAD. I’M NOT TAKING ANOTHER GENERIC UNTIL THEY START LIVE-STREAMING INSPECTIONS. THIS ISN’T SAFETY. THIS IS RUSSIAN ROULETTE WITH PILL BOTTLES.